iTol.py¶
Create files appropriate for use in the iTOL visualization program by using the abundance data from a biom-format file and groups specified in a QIIME mapping file. The program also modifies a Newick-format phylogenetic tree file to use proper taxonomic names instead of OTU IDs for useful display in iTOL.
usage: iTol.py [-h] -i OTU_TABLE -m MAPPING [-t INPUT_TREE] [-e OUTPUT_TRE] [-o OUTPUT_ITOL_TABLE] [-c MAP_CATEGORIES] [-a {MRA,NMRA,raw}]
Required arguments¶
-
-iOTU_TABLE,--otu_tableOTU_TABLE¶ The biom-format file with OTU-Sample abundance data.
-
-mMAPPING,--mappingMAPPING¶ The mapping file specifying group information for each sample.
Optional arguments¶
-
-tINPUT_TREE,--input_treeINPUT_TREE¶ A phylogenetic tree in Newick format to be modified by exchanging the OTU ID node names for taxonomic names.
-
-eOUTPUT_TRE,--output_treOUTPUT_TRE¶ The output Newick-format tree (.tre) file
-
-oOUTPUT_ITOL_TABLE,--output_itol_tableOUTPUT_ITOL_TABLE¶ Other than a phylogenetic tree, the main input to iTOL is a dataset file containing some representation of the abundance of every OTU across the specified data groups. This program provides multiple calculation methods. See the –analysis_metric option for details.
-
-cMAP_CATEGORIES,--map_categoriesMAP_CATEGORIES¶ Any mapping categories, such as treatment type, that will be used to group the data in the output iTOL table. For example, one category with three types will result in three data columns in the final output. Two categories with three types each will result in six data columns. Default is no categories and all the data will be treated as a single group.
-
-a{MRA,NMRA,raw},--analysis_metric{MRA,NMRA,raw}¶ Specifies which metric is calculated on the abundance data in the OTU table. Available options: MRE - mean relative abundance (Abundance data is normalized by total sample abundance, then averaged across OTU), NMRE - normalized mean relative abundance (MRE normalized by the total MRE across the groups as specified in –map_categories), raw (outputs the actual sequence abundance data for each OTU).
-
--stabilize_variance¶ Apply the variance-stabilizing arcsine square root transformation to the OTU proportion data. Recommended for usage with
-a NMRAor-a MRA.
-
-h,--help¶ Show the help message and exit.
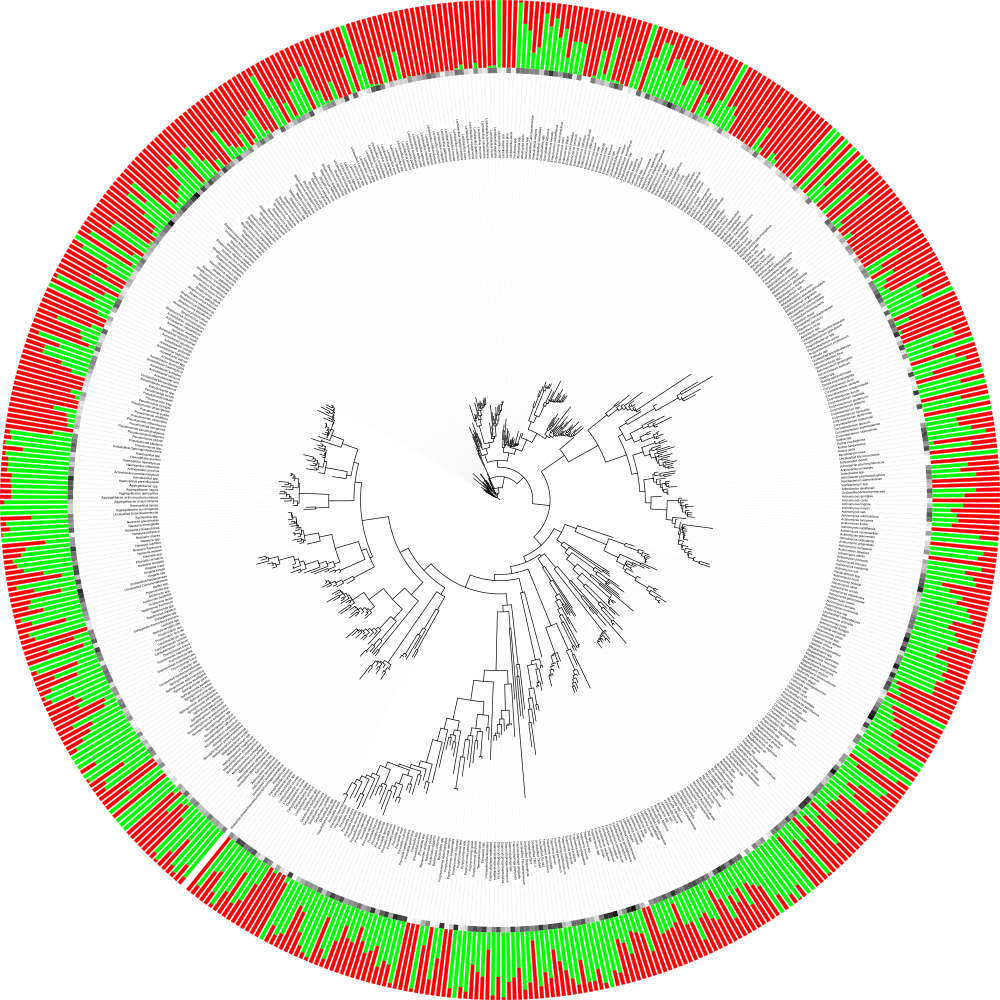
Workflow for generating useful phylogenetic trees using PhyloToAST¶
Step 1 : Obtain .tre file from QIIME’s make_phylogeny.py script.
Step 2 :
Run iTol.py script with -a NMRA analysis metric. This file will denote
the multibar graph around the circular phylogenetic tree.
Step 3 :
Run iTol.py script with -a raw analysis metric. This file will denote
the gradient graph around the circular phylogenetic tree.
Step 4 :
Upload modified .tre file from iTol.py script to iTOL website.
Add your dataset files and obtain the final phylogenetic tree figure.